
World Hemophilia Day 2024: The Invisible Reality of a Bleeding Disorder
This condition arrives without announcement. There are no visible marks, no obvious signs—just blood that refuses to stop flowing when it should. For millions worldwide living with hemophilia, this invisible reality shapes every aspect of existence, from childhood activities to menstrual cycles, from minor injuries to surgical procedures.
A Global Spotlight on an Overlooked Condition
Every year on April 17, World Hemophilia Day brings attention to this often-misunderstood disorder. The World Federation of Haemophilia (WFH) established this date to push governments and policymakers toward what seems deceptively simple: improved treatment, better access, and enhanced care. The fact that this advocacy remains necessary decades after the day's establishment reveals how quietly hemophilia continues to be sidelined in healthcare discussions globally.
At its fundamental level, hemophilia represents a clotting disorder. The body lacks or has insufficient levels of specific proteins known as Factor VIII or Factor IX, which signal blood to cease flowing after injury. Without these crucial proteins, even internal bleeding into joints or muscles can become life-threatening. This rare, inherited condition remains widely misunderstood despite medical advancements.

The Genetic Reality and Gender Myths
The condition maintains deep genetic connections. Because hemophilia links to the X chromosome, men face significantly higher diagnosis probabilities. A boy born to a carrier mother carries a 50 percent chance of inheriting the condition. However, the persistent myth that hemophilia "only affects men" quietly erases millions of women who carry the gene and confront their own unique risks, including heavy menstrual periods, childbirth complications, and unexpected bleeding episodes.
This erasure represents precisely what World Hemophilia Day attempts to address—not merely in policy frameworks but in public perception and medical understanding.
The Daily Reality: Pain, Precautions, and Bleeding Risks
Most people visualize hemophilia as a problem with external cuts—someone bleeds and cannot stop. The reality proves both less dramatic and far more serious than this common perception.
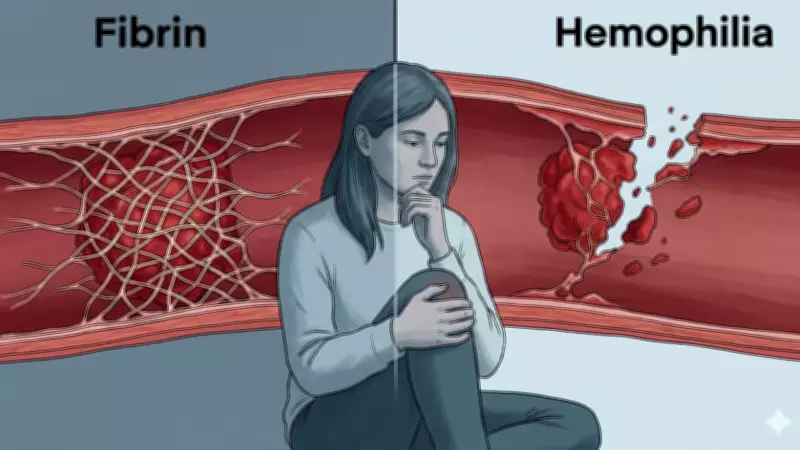
Here's what actually occurs during normal clotting: First, injured blood vessels contract to slow blood flow. Next, platelets swarm the area, forming a temporary plug. Finally, clotting proteins weave a fibrin mesh over this plug, creating a tight seal that holds wounds closed during healing.
For individuals with hemophilia, the first two steps typically function adequately. Blood vessels contract, and platelets arrive. However, the third step involving fibrin clot formation either fails completely or produces such thin, fragile clots that they collapse. Bleeding doesn't stop, or it pauses briefly before resuming.
This explains why the common assumption—that hemophilia patients bleed to death from paper cuts—proves largely incorrect yet not entirely reassuring. Small surface injuries rarely pose danger. The real threat resides deeper within the body.
When Bleeding Occurs Internally
Internal bleeding transforms hemophilia into a life-altering condition. Blood pooling inside joints or muscles creates pressure and damage. Knees, elbows, and ankles represent the most common targets. Over time, repeated bleeding into joints—even when treated—breaks down cartilage, stiffens tissues, and causes arthritis decades earlier than typical.
What makes management particularly challenging involves how ordinary the triggers appear. A bumped knee during morning commutes, an elbow knocked against a doorframe—injuries so minor that unaffected individuals wouldn't register them. For some patients, no trigger exists at all; bleeding begins mid-stride during otherwise unremarkable days. These "spontaneous bleeds" arrive unpredictably, uninvited, and urgently.
Timing proves critical here. Unlike cuts addressable with bandages, internal bleeds require rapid treatment with clotting factor infusions before pressure and damage compound. Delayed recognition or treatment means increased pain, greater joint damage, and prolonged recovery periods.

This constitutes the daily arithmetic of living with hemophilia: measuring risk against routine, reading bodily signals for early warnings, and carrying the knowledge that ordinary days can transform into complex medical situations rapidly.
"Patients suffering from hemophilia face multiple levels of daily challenges," explains Dr. Geetika Jassal, Medical Spokesperson at Cryoviva Life Sciences. "One primary issue involves constant spontaneous bleeding risks, especially into joints, leading to chronic pain, swelling, and reduced mobility over time. Managing the condition requires regular clotting factor infusions, frequent hospital visits, and strict treatment adherence—physically exhausting and financially burdensome."
From Diagnosis to Delay: Global Detection Challenges
For a condition documented for centuries, hemophilia remains remarkably easy to miss. Reasons appear layered. Hemophilia exists on a spectrum—severe, moderate, and mild—with each level behaving differently enough to resemble three separate conditions.
Severe hemophilia may show early childhood signs: unexplained bruising, bleeding after minor falls, swollen joints without apparent cause. Mild hemophilia patients might continue for years—sometimes decades—without obvious episodes. Their blood contains just enough clotting factor activity to handle everyday scrapes. Only during significant events like surgeries, tooth extractions, serious accidents, or childbirth does the body's quiet insufficiency finally surface, often delivering diagnosis as a shocking revelation.
The Family History Gap
Many families request newborn testing when hemophilia runs in their lineage—a reasonable precaution catching significant cases early. However, approximately one in three babies diagnosed with hemophilia carries a new genetic mutation absent in both parents. No family history exists, no reason to investigate, just results arriving without warning after something has already gone wrong.
This means for a substantial portion of hemophilia patients, the condition never appeared on anyone's radar until it announced itself through medical crisis.
"Early hemophilia indications usually appear subtle, often mistaken for normal childhood injuries," Dr. Jassal adds. "These include frequent bruising, prolonged bleeding from minor cuts, recurrent nosebleeds, and excessive bleeding after injections, vaccinations, or dental procedures. In infants particularly, joint swelling presenting as irritability or reduced limb movement may go untreated. Many parents dismiss these signs as normal, delaying diagnosis. Since symptoms aren't always dramatic early on, hemophilia often gets misdiagnosed or identified only after major bleeding events or surgical complications."
The Overlooked Women
Then exists the most consistently overlooked group: women. The longstanding assumption that hemophilia represents a male disease has quietly shaped how doctors ask questions and how women understand their own symptoms.
A woman carrying the hemophilia gene might experience heavy menstrual cycles continuing too long, childbirth bleeding complications, or wounds healing slower than normal. These represent real symptoms with real causes. Without appropriate questions or awareness, they risk misdiagnosis or complete dismissal. Diagnosis sometimes follows years of being told nothing is wrong.
What Detection Actually Involves
When hemophilia suspicion arises, the process proves straightforward: blood tests check whether clotting occurs properly, and factor assays identify missing proteins and severity levels. Tests and knowledge exist. The gap rarely involves medicine itself but rather whether anyone considered investigating initially.
This gap widest in low-income countries where specialist care access remains limited and general practitioner awareness uneven. Yet it persists even in well-resourced healthcare systems—quietly, in mild cases never triggering alarm, in women whose symptoms were explained away, in newborns with unannounced mutations.
Early detection significantly changes outcomes, allowing families to prepare, doctors to intervene before joint damage sets in, and individuals to understand their bodies before crises force conversations. Medicine has advanced; awareness in too many places still catches up.
"Early hemophilia diagnosis proves crucial to prevent irreversible joint damage, life-threatening bleeding episodes, and long-term disability," emphasizes Dr. Jassal. "Identifying the condition early allows prophylactic clotting factor therapy initiation, significantly improving quality of life and long-term outcomes. However, significant awareness gaps persist and remain major concerns. Early signs like prolonged bleeding after minor injuries, easy bruising, or joint swelling often get overlooked or misdiagnosed, especially by primary healthcare providers."
From Plasma Bags to Precision Medicine: Treatment Evolution
In 1964, when first effective hemophilia treatments took shape, options remained basic. Cryoprecipitate—frozen plasma concentrate—represented the best available. For most patients, treatment meant reaching one of few cities with proper equipment, hoping for supply availability, and managing everything else through caution and luck.
The distance between that world and today's reality appears enormous. Hemophilia treatment has progressed from plasma-derived products to recombinant clotting factor concentrates, and more recently to extended half-life therapies reducing infusion frequency. Where patients once required intravenous clotting factor injections multiple times weekly, newer therapies permit less frequent administration. Some can be delivered subcutaneously rather than intravenously—a significant difference for children and those living far from medical facilities.
Then arrived emicizumab, a bispecific antibody functioning differently from traditional factor replacement altogether. Administered subcutaneously with less frequent dosing, it doesn't trigger inhibitor development that undermines traditional factor concentrates in some patients—treatment that would have seemed implausible a generation ago.
Now at the frontier: gene therapy. Unlike all previous treatments, gene therapy requires only single administration, prompting the liver itself to produce Factor VIII or IX—the body performing what it never could before. The FDA approved two gene therapies for hemophilia B (Hemgenix in 2022 and Beqvez in 2024) and one for severe hemophilia A. These represent not incremental improvements but an entirely different medicine category.
"Recent hemophilia studies explore advanced approaches like gene therapy and stem cell-based strategies, mainly addressing root causes rather than just managing symptoms," notes Dr. Jassal. "Stem cell research remains largely experimental, with ongoing studies evaluating its potential for sustained clotting factor production. While these developments hold long-term promise, they aren't yet part of routine clinical practice."
Where Does India Stand?
India carries the world's second-largest hemophilia burden, with approximately 136,000 estimated cases. Yet the country that recently completed its first in-human gene therapy trial for the condition also sees most patients managing bleeds with rest, ice, and whatever else they can find. This contradiction sits at the heart of Indian hemophilia care today: world-class science at one extreme, and a vast, underserved majority at the other.
The Insurance and Financial Problem
Hemophilia occupies an awkward position in India's healthcare economy—too rare for sustained policy attention, too expensive to treat without it. Private health insurance remains largely inaccessible for most affected families, and hemophilia's classification as a low-volume, high-cost disease means insurers possess little incentive for adequate coverage.
The result: economic circumstances dictate treatment in ways they shouldn't. For many patients, factor replacement therapy remains something they read about rather than receive. Instead, they manage with RICE (rest, ice, compression, elevation), alongside adjunct medications and wet products like fresh frozen plasma or cryoprecipitate when available. These don't constitute treatments so much as daily survival methods.
"In India, access remains very limited to select research settings and specialized centers, with current standard care still centered around clotting factor replacement and new non-factor therapies when available," states Dr. Jassal.
The overall picture reveals a system still treating a chronic condition as a series of emergencies. Untreated hemophilia costs—hospitalizations, surgeries, long-term disability, lost productivity—ultimately burden families and health systems, burdens preventable through early, consistent care.





